MASH
MASH is for uncovering metagenomic distances using MinHash
Parameters
FASTA genome file: file
Whatever genome file you want to uncover genomic distances from. Can be multipe entries. Can also be a consensus file
- Reference Genomes: File
Can be Sketch format
.mshor FASTA file.
Sketch File 1: option
Speeds up the process for the input file 1
Sketch File 2: option
Speeds up the process for the input file 2
Winnder Take All: option
Only take the highest (best) distance annotations against the reference for your input file.
Returns

Distance: tab-separated file
Note
- Columns:
Reference-ID
Query-ID
Mash-distance
P-value
Matching-hashes
See more information on calculaions here
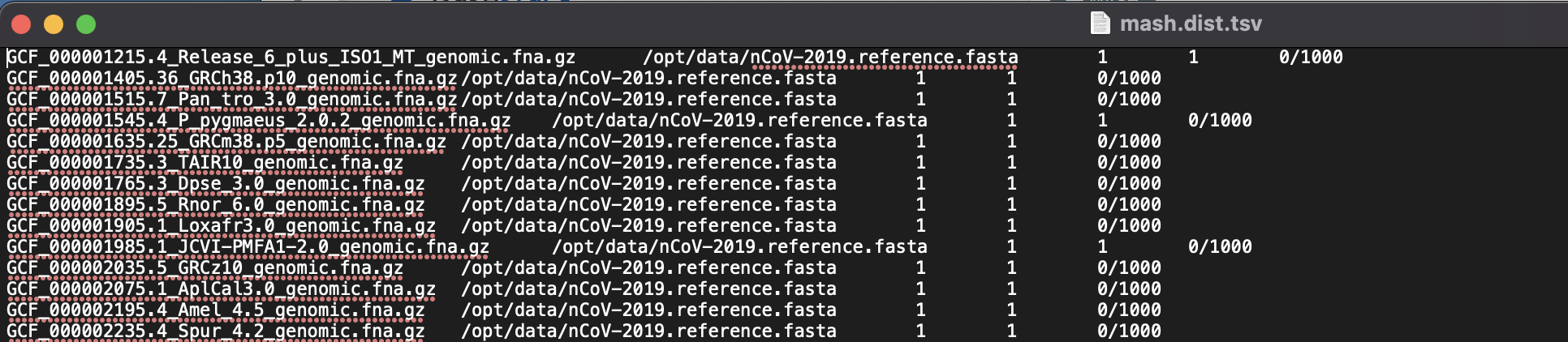
Screen: tab-separated file
Note
- Columns:
identity
shared-hashes
median-multiplicity
p-value
query-ID
query-comment
See more information here
