Artic Field Bioinformatics
Artic is a tool designed for analyzing and creating consensuses from viral nanopore sequencing reads. These also are tied to the use of amplicon schemes (tiled)
Note
This module will output multiple consensuses per barcode of interest. It takes in a raw fastq_pass directory and outputs .vcfs, .fasta, and .bam files for further use
Medaka
Parameters
input run folder: directory
Must contain a
fastq_passdirectory or a custom inputted one (See below)Primer: option or Directory
Artic Primer set or a custom one (directory) which contain the
genome.fastaand necessaryBEDfile for the primer setNormalize Coverage: Number
Barcode Configuration: option
Which barcode kit you used for demux. Select any for non-barcoded sample
FASTQ Dir: Directory, optional
Select your own custom fastq_pass directory to analyze and demux
Medaka Model: option
FAST or HAC used during basecalling
Returns
Medaka Consensus files: FASTA files containing your consensuses for each barcode
Medaka VCF files: Variant files containing your variant calls for each barcode
Medaka BAM files: BAM files containing your alignment information for each barcode


Nanopolish
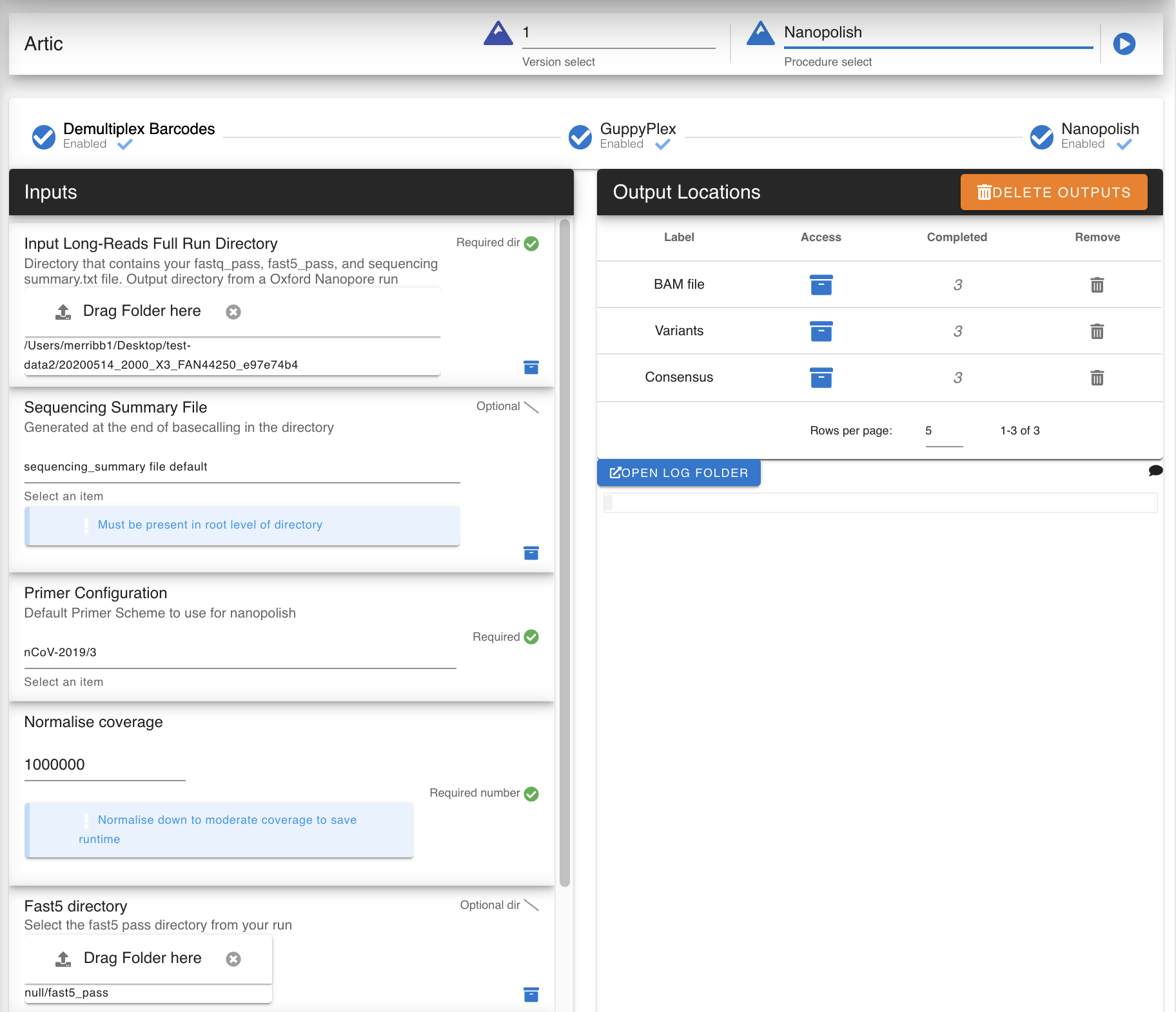

Parameters
input run folder: directory
Must contain a
fastq_passdirectory or a custom inputted one (See below)Primer: option or Directory
Artic Primer set or a custom one (directory) which contain the
genome.fastaand necessaryBEDfile for the primer setNormalize Coverage: Number
Barcode Configuration: option
Which barcode kit you used for demux. Select any for non-barcoded sample
FASTQ Dir: Directory, optional
Select your own custom fastq_pass directory to analyze and demux
Sequencing Summary File: File, exists
Selects the Sequencing summary file in the root of your run directory
Note
Oftentimes, this file can be found in the
fastq_passdirectory. Move it one level up to the root run directory
Returns
Nanopolish Consensus files: FASTA files containing your consensuses for each barcode
Nanopolish VCF files: Variant files containing your variant calls for each barcode
Nanopolish BAM files: BAM files containing your alignment information for each barcode